-
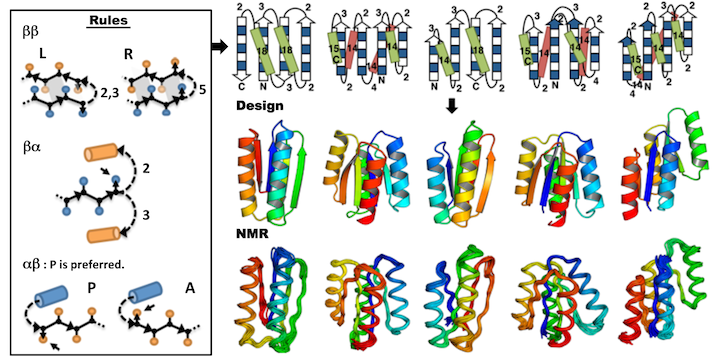
PRINCIPLES FOR DESIGNING IDEAL PROTEIN STRUCTURES
Koga, N., Tasumi-Koga R., et al., Nature. 491(7423), 222-227. (2012) We describe an approach to designing ideal protein structures stabilized by completely consistent local and non-local interactions. The approach is based on a set of rules relating secondary structure patterns to protein tertiary motifs, which make possible the design of strongly funneled protein folding energy…
-
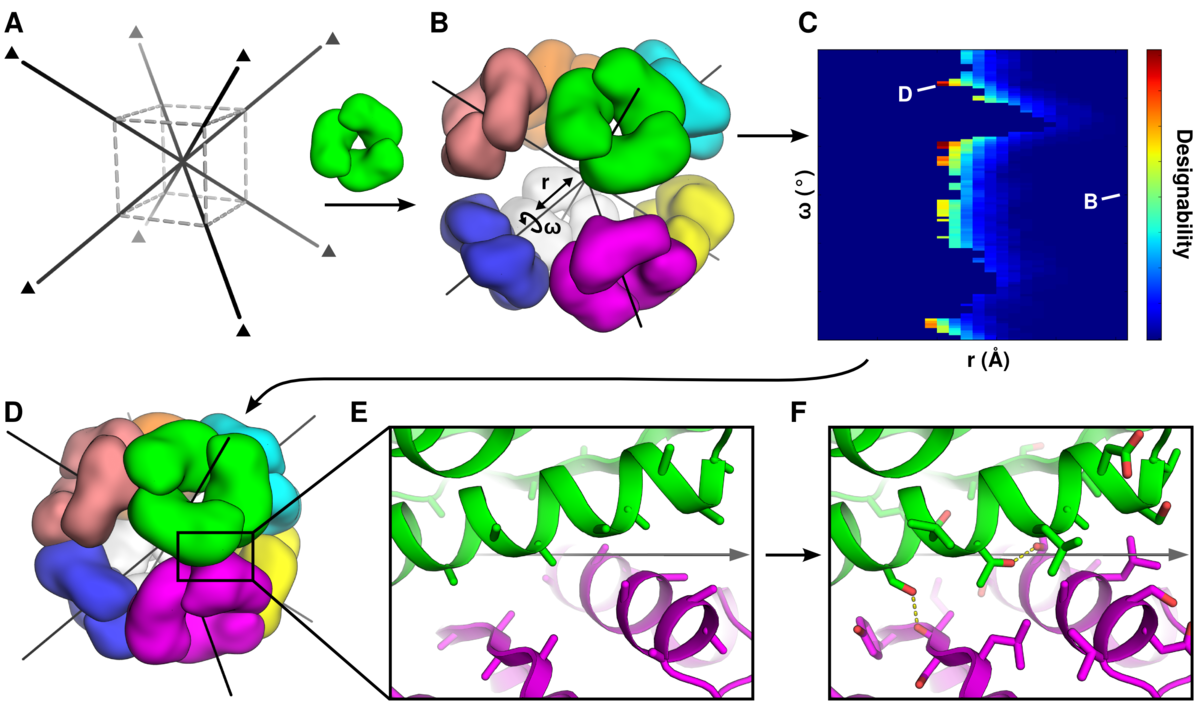
Computational Design of Self-Assembling Protein Nanomaterials with Atomic Level Accuracy
King, N.P., Sheffler, W., et al. Science. 336(6085), 1171-1174. (2012) We describe a general computational method for designing proteins that self-assemble to a desired symmetric architecture. Protein building blocks are docked together symmetrically to identify complementary packing arrangements, and low-energy protein-protein interfaces are then designed between the building blocks in order to drive self-assembly. Here…
-
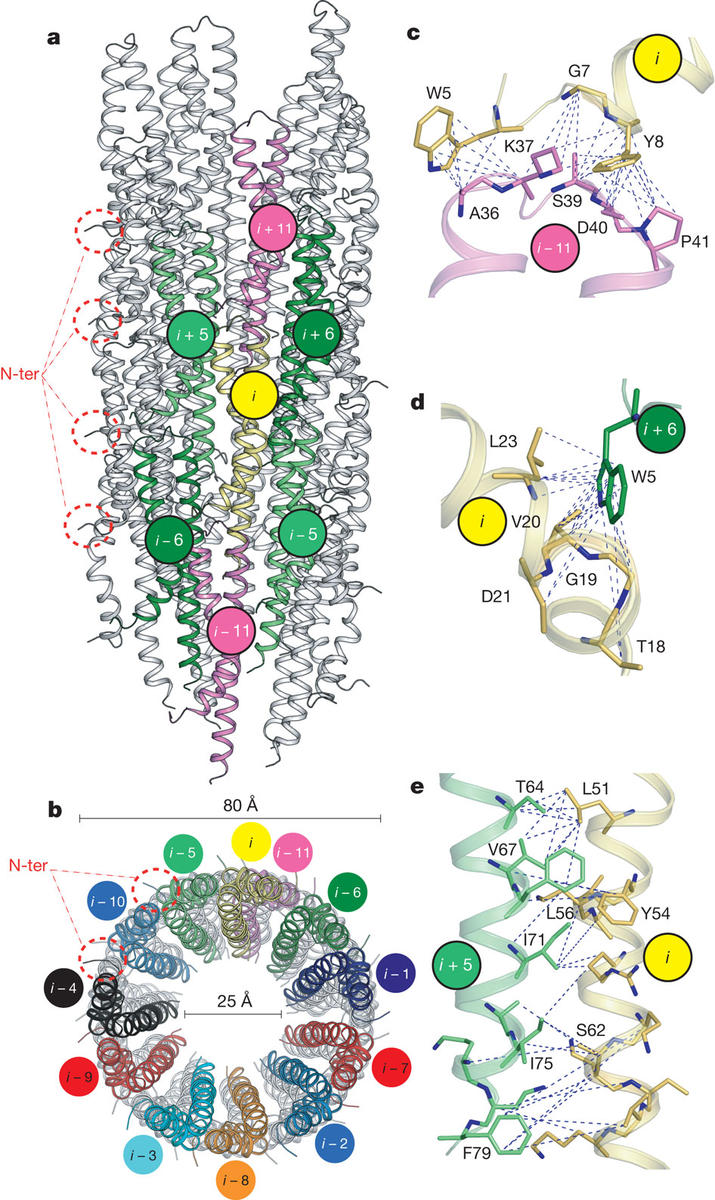
Atomic model of the type III secretion system needle
Loquet, A., Sgourakis N. G., et al. Nature (2012) The ability of Gram-negative bacteria, such as the agents of plague, dysentery and typhoid fever to infect host cells is dependent on a syringe-like molecular machine known as the Type-III secretion system (T3SS). The core of T3SS consists of a hollow filament, the needle; composed of…
-
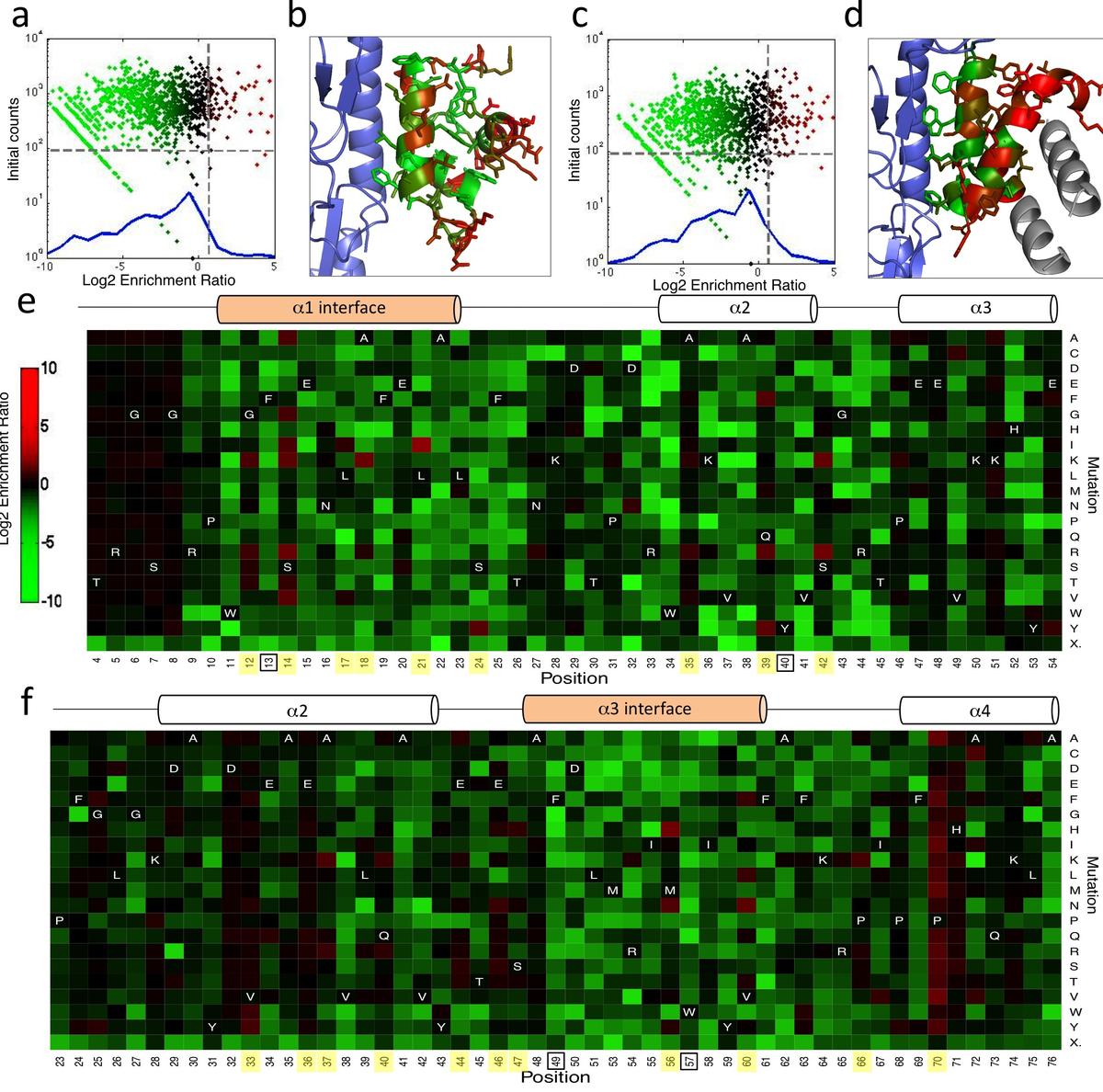
Optimization of affinity, specificity and function of designed influenza inhibitors using deep sequencing
Whitehead, T. A., Chevalier A. et al. Nature biotechnology (2012) We show that comprehensive sequence-function maps obtained by deep sequencing can be used to reprogram interaction specificity and to leapfrog over bottlenecks in affinity maturation by combining many individually small contributions not detectable in conventional approaches. We use this approach to optimize two computationally designed…
-
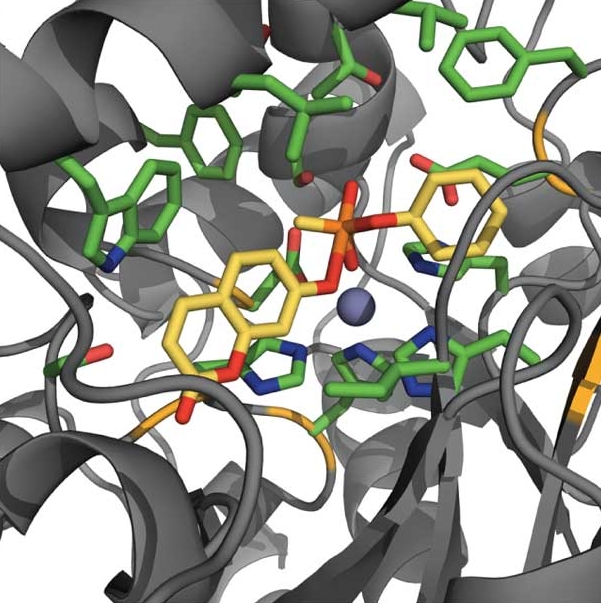
Computational redesign of a mononuclear zinc metalloenzyme for organophosphate hydrolysis
Khare, S. D., Kipnis Y., et al. Nature chemical biology (2012) The ability to redesign enzymes to catalyze noncognate chemical transformations would have wide-ranging applications. We developed a computational method for repurposing the reactivity of metalloenzyme active site functional groups to catalyze new reactions. Using this method, we engineered a zinc-containing mouse adenosine deaminase to…
-
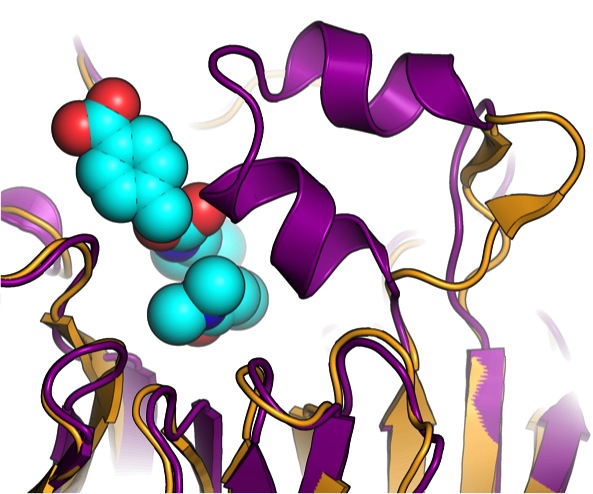
Increased Diels-Alderase activity through backbone remodeling guided by Foldit players
Eiben, C. B., Siegel J. B., Bale J. B. et al. Nature biotechnology. 30(2), 190-2. (2012) Computational enzyme design holds promise for the production of renewable fuels, drugs and chemicals. De novo enzyme design has generated catalysts for several reactions, but with lower catalytic efficiencies than naturally occurring enzymes. Here we report the use of game-driven crowdsourcing to enhance the activity…
-
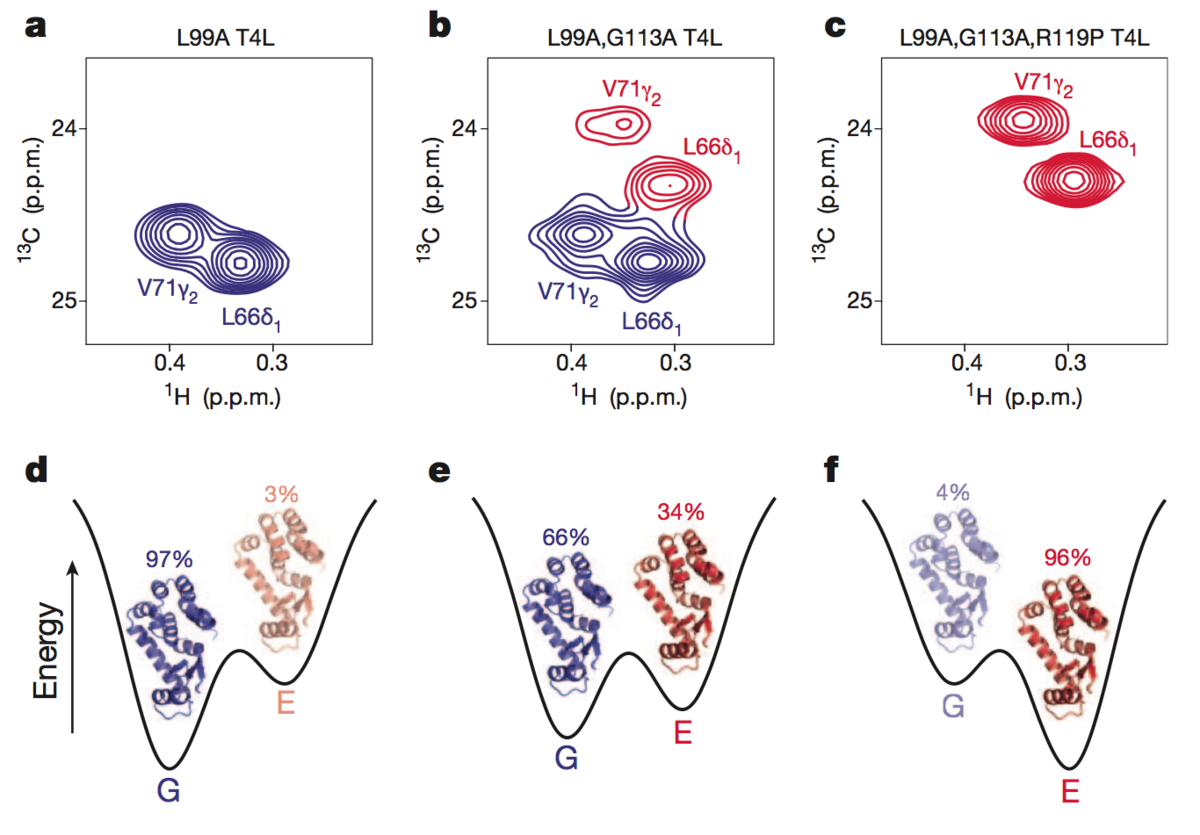
Solution structure of a minor and transiently formed state of a T4 lysozyme mutant
Bouvignies, G., Vallurupalli P., et al. Nature. 477(7362), 111-4. (2011) Proteins are inherently plastic molecules, whose function often critically depends on excursions between different molecular conformations (conformers). However, a rigorous understanding of the relation between a protein’s structure, dynamics and function remains elusive. This is because many of the conformers on its energy landscape are only transiently formed…
-
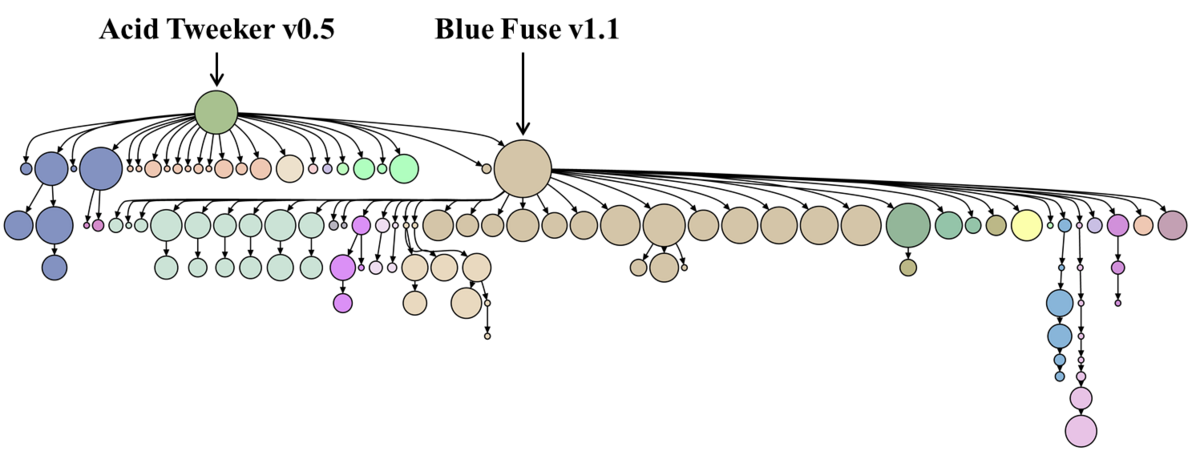
Algorithm discovery by protein folding game players
Khatib, F., Cooper S., Tyka M. D., Xu K., Makedon I., Popovic Z., et al. Proc Natl Acad Sci USA (2011) To determine whether high performing Foldit player strategies could be collectively codified, we augmented the Foldit gameplay mechanics with tools for players to encode their folding strategies as “recipes” and to share their recipes…