Tag: Peilong Lu
-
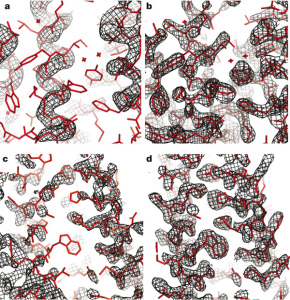
High Resolution Protein Structure Refinement
Qian B, Raman S, et al. Nature 450, 259-64. (2007) A longstanding problem in computational biology is the refinement of low resolution protein structure models to more atomic-level accurate structures. A related challenge is refining low-resolution NMR models to the quality of high-resolution structures. NMR is a valuable tool for determining protein structures, particularly because…
-
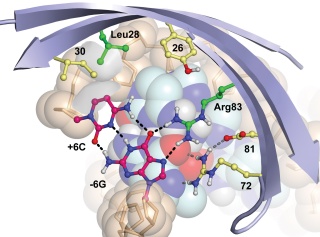
Computational Design of Protein-DNA Cleavage Specificity in a Homing Endonuclease
Ashworth J, Havranek JJ, et al. Nature 441, 656-9. (2006) High-resolution modeling of protein-DNA interactions has granted us the ability to estimate the specificities of real and hypothetical interfaces. This approach may be useful to design novel sequence-specific endonucleases for biotechnology and medicine. The figure at left depicts the (crystallographically and biochemically validated) model of…
-
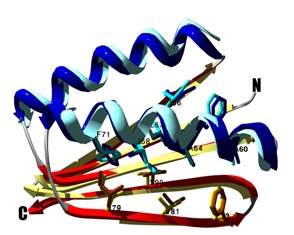
Design of a novel globular protein fold with atomic level accuracy
Kuhlman B, Dantas G, et al. Science 302, 1364-8. (2003) A major challenge of computational protein design is the creation of novel proteins with arbitrarily chosen three-dimensional structures. Here, we used a general computional strategy that iterates between sequence design and structure prediction to design a 93-residue alpha/beta protein called Top7 with a novel sequence…
-
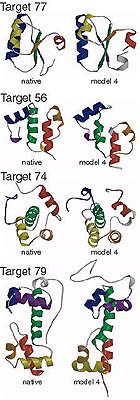
Blind ab initio structure prediction of CASP3 targets
Simons, K.T., Bonneau, et al. Proteins 3, 171-176 (1999) To generate structures consistent with both the local and non-local interactions responsible for protein stability, 3 and 9 residue fragments of known structures with local sequences similar to the target sequence were assembled into complete tertiary structures using a Monte Carlo simulated annealing procedure (Simons, K.T.…
-
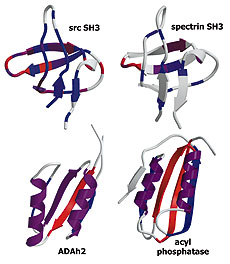
Experiment and theory highlight role of native state topology in SH3 folding
Riddle, D.S., Grantcharova, V.P., et al. Nat Struct Biol 6, 1016-1204. (1999) We use a combination of experiments, computer simulations and simple model calculations to characterize, first, the folding transition state ensemble of the src SH3 domain, and second, the features of the protein that determine its folding mechanism. Kinetic analysis of mutations at 52…
-
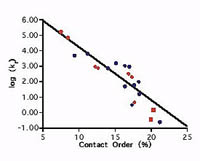
A correlation between folding rate and contact order
Plaxco, K. W., Simons, K. T. et al. J. Mol. Biol. 277, 985-994. (1998) Our studies have revealed a significant correlation between the average sequence seqaration between contacting residues in the native state (contact order) and the folding rate of simple, single domain proteins. Calculate the contact order for your protein or a protein in…
-
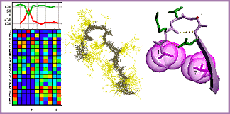
The I-sites library of sequence-structure motifs
Bystroff, C. and Baker, D. J. Mol. Biol. 281, 565-77. (1998) I-Sites Library Homepage I-Sites Prediction Server I-Sites clusters by motif
-
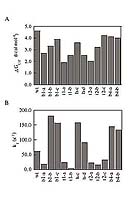
Sequences of small proteins are not optimized for rapid folding
Kim, D. E., Gu, H., and Baker, D. Proc. Natl. Acad. Sci, 95, 4982-4986 (1998) Distributions of free energies of unfolding and refolding rates in randomized protein L variants compared to wild type.