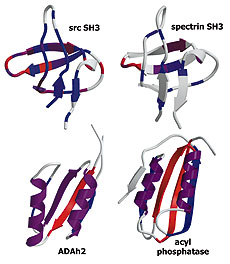
Riddle, D.S., Grantcharova, V.P., et al. Nat Struct Biol 6, 1016-1204. (1999)
We use a combination of experiments, computer simulations and simple model calculations to characterize, first, the folding transition state ensemble of the src SH3 domain, and second, the features of the protein that determine its folding mechanism. Kinetic analysis of mutations at 52 of the 57 residues in the src SH3 domain revealed that the transition state ensemble is even more polarized than suspected earlier: no single alanine substitution in the N-terminal 15 residues or the C-terminal 9 residues has more than a two-fold effect on the folding rate, while such substitutions at 15 sites in the central three-stranded beta-sheet cause significant decreases in the folding rate. Molecular dynamics (MD) unfolding simulations and ab initio folding simulations on the src SH3 domain exhibit a hierarchy of folding similar to that observed in the experiments. The similarity in folding mechanism of different SH3 domains and the similar hierarchy of structure formation observed in the experiments and the simulations can be largely accounted for by a simple native state topology-based model of protein folding energy landscapes.
Related papers:
| Goldenberg, D. P. (1999). Finding the right fold. Nat Struct Biol 6, 987-990. | |
| Chiti, F., Taddei, N., White, P. M., Bucciantini, M., Magherini, F., Stefani, M., and Dobson, C. M. (1999). Mutational analysis of acylphosphatase suggests the importance of topology and contact order in protein folding. Nat Struct Biol 6, 1005-1009. | |
| Martinez, J. C. and Serrano, L. (1999). The folding transition state between SH3 domains is conformationally restricted and evolutionarily conserved. Nat Struct Biol 6, 1010-1016. |