-
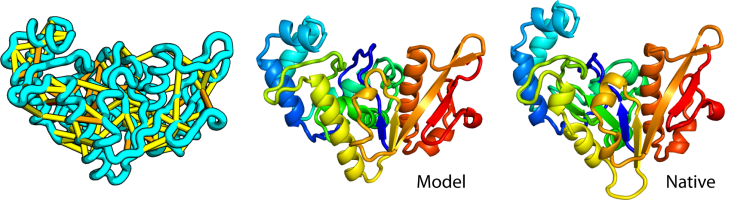
Large-scale determination of previously unsolved protein structures using evolutionary information
The prediction of the structures of proteins without detectable sequence similarity to any protein of known structure remains an outstanding scientific challenge. Here we report significant progress in this area. We first describe de novo blind structure predictions of unprecendented accuracy we made for two proteins in large families in the recent CASP11 blind test…
-

Design of ordered two-dimensional arrays mediated by noncovalent protein-protein interfaces
We describe a general approach to designing two-dimensional (2D) protein arrays mediated by noncovalent protein-protein interfaces. Protein homo-oligomers are placed into one of the seventeen 2D layer groups, the degrees of freedom of the lattice are sampled to identify configurations with shape-complementary interacting surfaces, and the interaction energy is minimized using sequence design calculations. We…
-
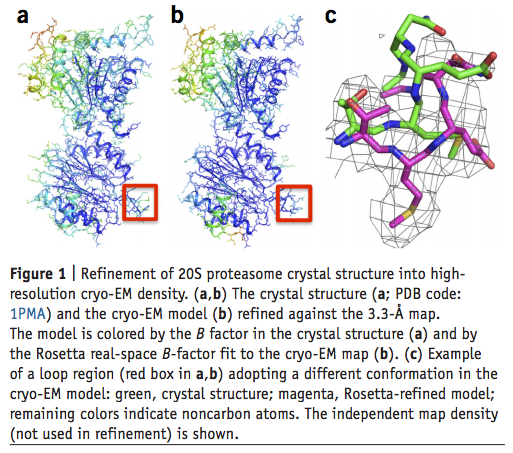
Atomic-accuracy models from 4.5-Å cryo-electron microscopy data with density-guided iterative local refinement
We describe a general approach for refining protein structure models on the basis of cryo-electron microscopy maps with near-atomic resolution. The method integrates Monte Carlo sampling with local density-guided optimization, Rosetta all-atom refinement and real-space B-factor fitting. In tests on experimental maps of three different systems with 4.5-Å resolution or better, the method consistently produced…
-
Trapping a transition state in a computationally designed protein bottle
The fleeting lifetimes of the transition states (TSs) of chemical reactions make determination of their three-dimensional structures by diffraction methods a challenge. Here we used packing interactions within the core of a protein to stabilize the planar TS conformation for rotation around the central carbon-carbon bond of biphenyl so that it could be directly observed…
-
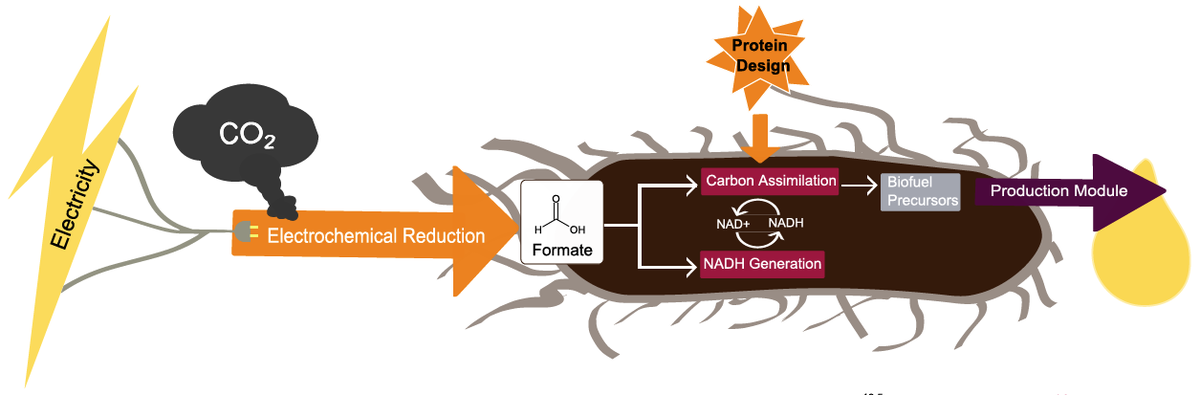
Computational protein design enables a novel one-carbon assimilation pathway
We describe a computationally designed enzyme, formolase (FLS), which catalyzes the carboligation of three one–carbon formaldehyde molecules into one three-carbon dihydroxyacetone molecule. The existence of FLS enables the design of a new carbon fixation pathway, the formolase pathway, consisting of a small number of thermodynamically favorable chemical transformations that convert formate into a three-carbon sugar in central metabolism. The formolase pathway is predicted to use carbon more…
-
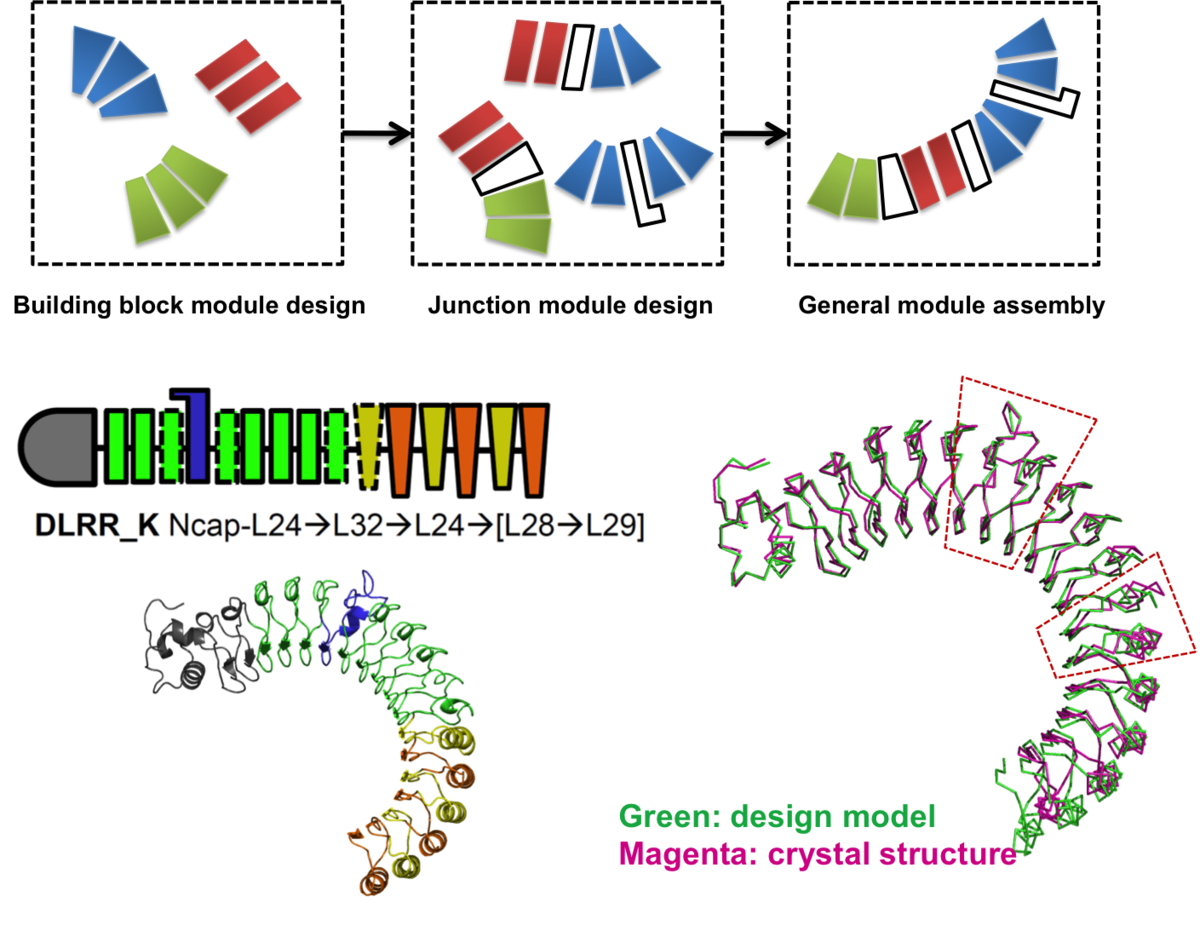
Control of repeat-protein curvature by computational protein design
Shape complementarity is an important component of molecular recognition, and the ability to precisely adjust the shape of a binding scaffold to match a target of interest would greatly facilitate the creation of high-affinity protein reagents and therapeutics. Here we describe a general approach to control the shape of the binding surface on repeat-protein scaffolds…
-
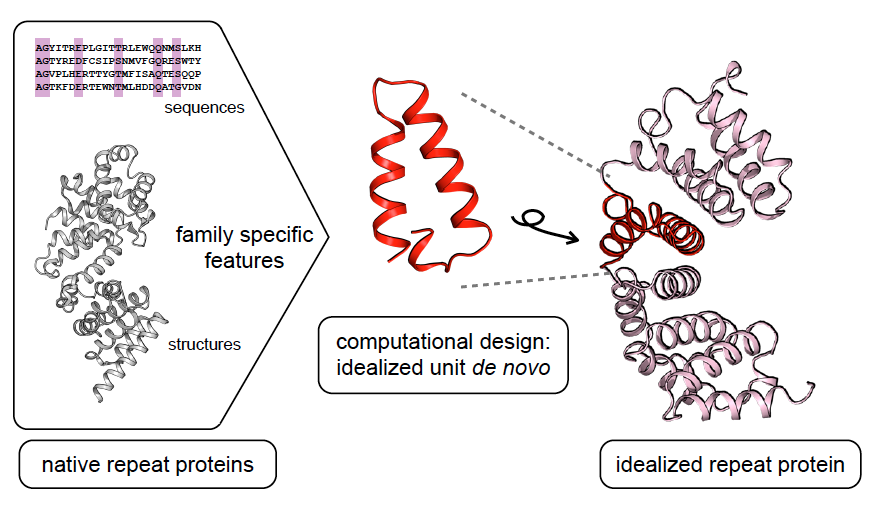
A general computational approach for repeat protein design
Repeat proteins have considerable potential for use as modular binding reagents or biomaterials in biomedical and nanotechnology applications. Here we describe a general computational method for building idealized repeats that integrates available family sequences and structural information with Rosetta de novo protein design calculations. Idealized designs from six different repeat families were generated and experimentally…
-

High thermodynamic stability of parametrically designed helical bundles
We describe a procedure for designing proteins with backbones produced by varying the parameters in the Crick coiled coil–generating equations. Combinatorial design calculations identify low-energy sequences for alternative helix supercoil arrangements, and the helices in the lowest-energy arrangements are connected by loop building. We design an antiparallel monomeric untwisted three-helix bundle with 80-residue helices, an…