Category: Uncategorized
-

Custom proteins help the body heal
New regenerative medicine proteins called NeoNectins help biomaterials work better with the body by guiding cells to attach, grow, and heal.
-

Congrats Roman!
Postdoc Roman Barth, PhD, has received a Harold M. Weintraub Graduate Student Award. He’s now building new molecular machines.
-
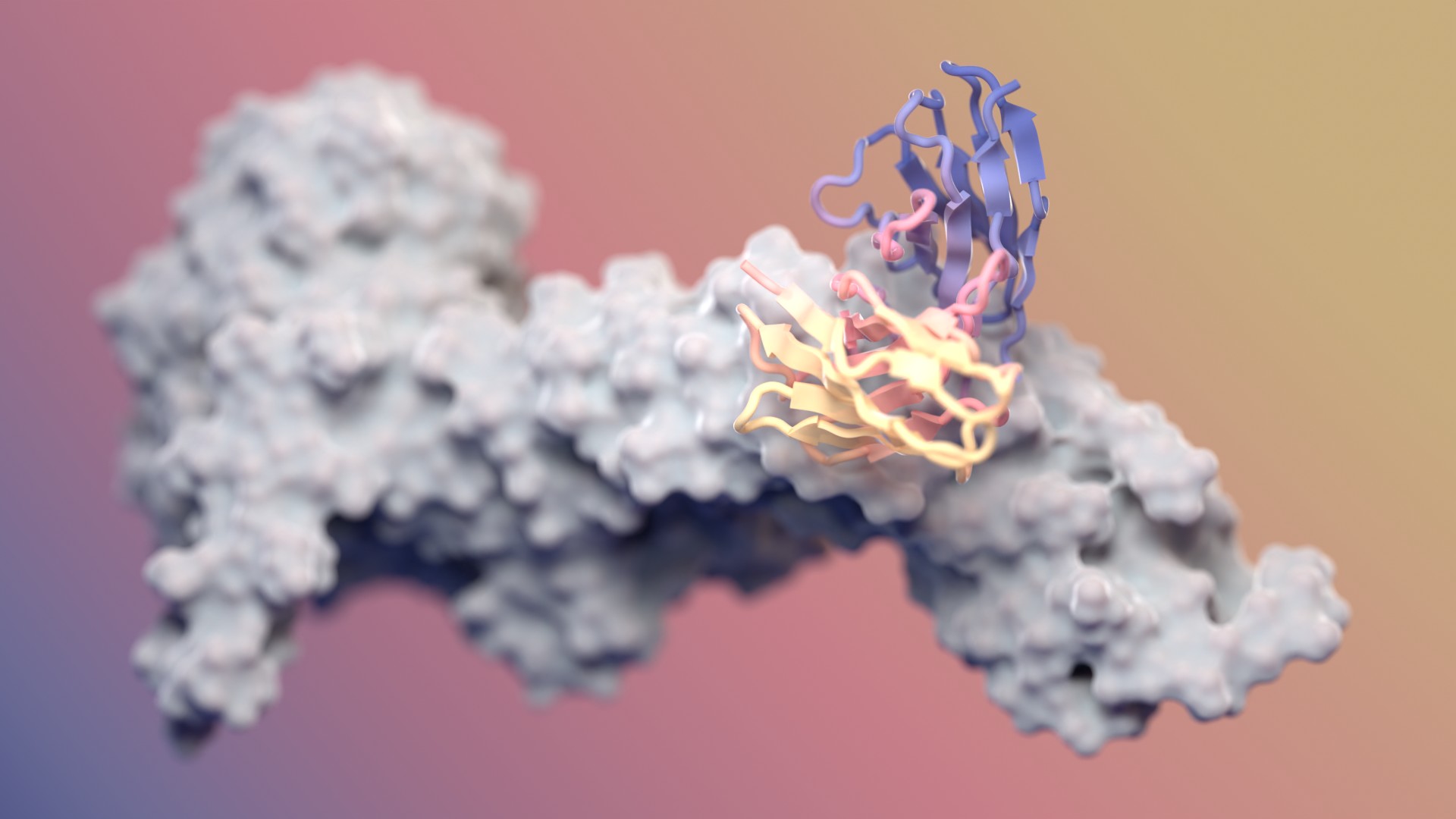
Designing antibodies with RFdiffusion
Our fine-tuned AI model generates functional antibodies with atomic precision. It’s also free to use.
-

Generating new enzymes with complex active sites
Custom catalysts may help break down plastics and synthesize medicines.
-
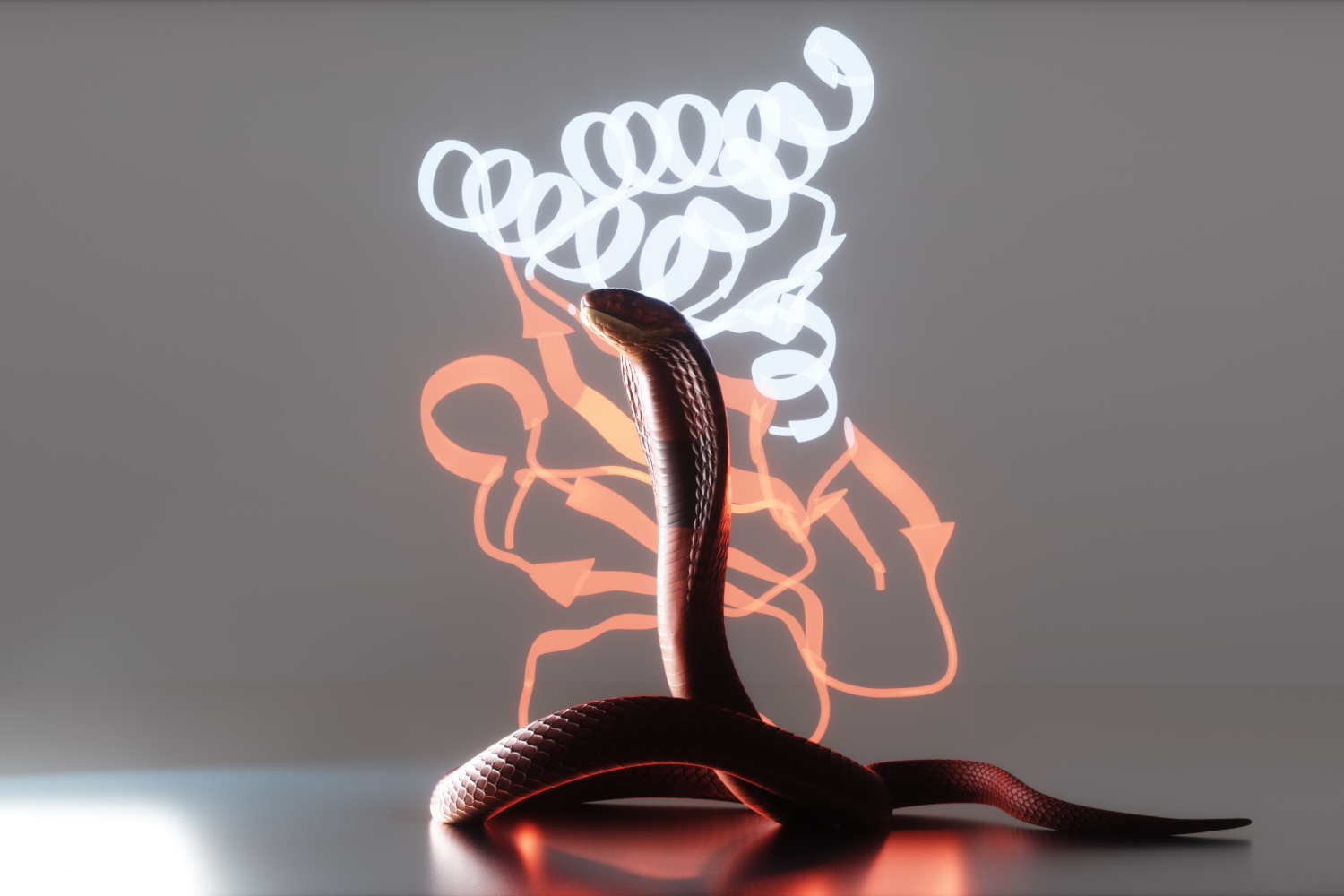
Neutralizing deadly snake toxins
AI-generated proteins that protect animals in the lab promise safer, more accessible antivenoms.
-

Congrats Han!
Postdoc Han Raut Altae-Tran, PhD, has received a Life Sciences Research Foundation fellowship. He’s designing RNA-binding proteins to regulate key biological functions.
-
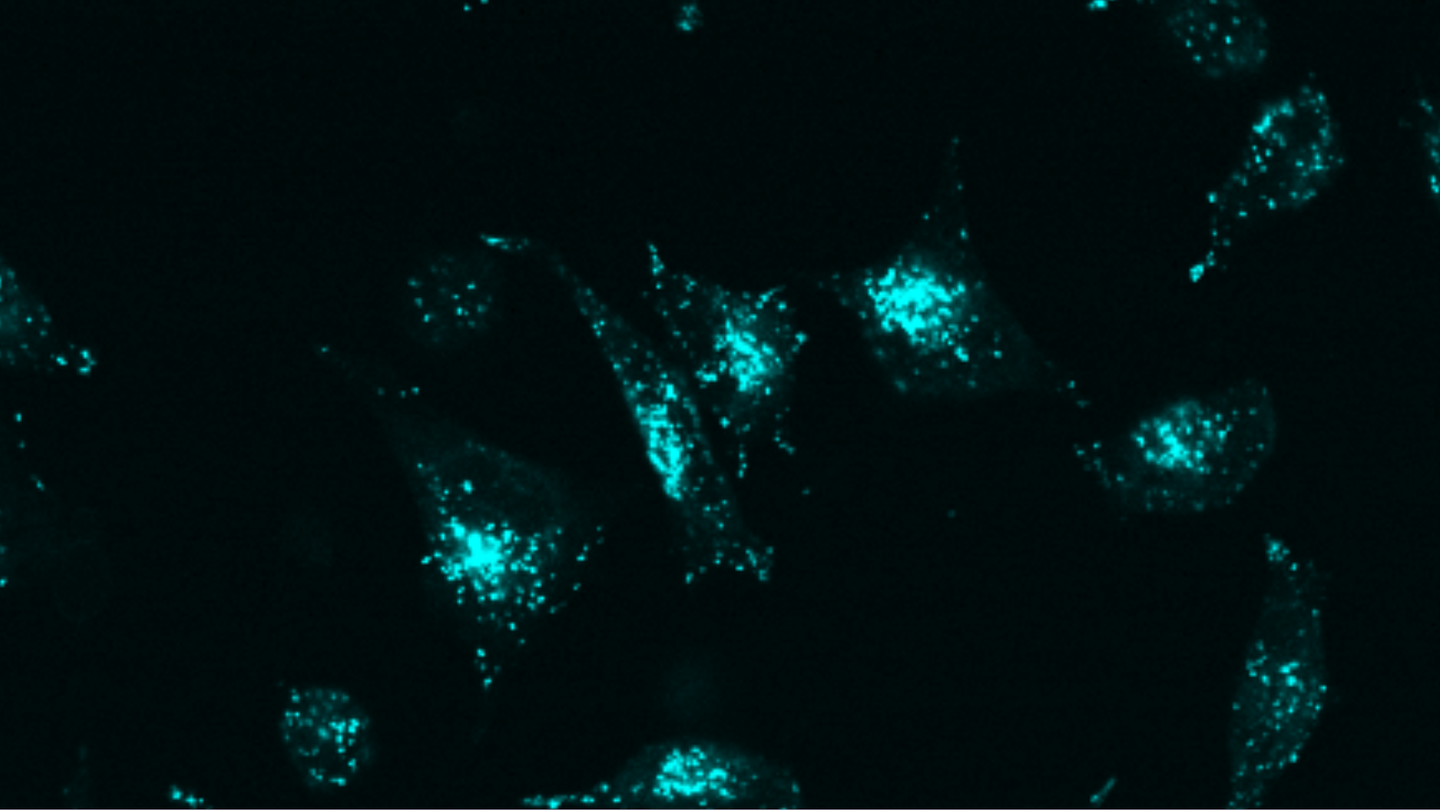
Targeted protein degradation through advanced protein design
“We’ve created a new approach that uses simpler components, achieves greater precision, and can be applied to virtually any target molecule on the surface of cells.”
-

Design of noncanonical alpha helices
We’ve found new secondary structures by screening over 100 uncommon amino acids, unlocking fresh building blocks for molecular engineering.