Jiang, Lin, Althoff Eric A. et al, Science, 319, 1387-91. (2008)
 Using new algorithms that employ hashing techniques to construct active sites for multi-step reactions, we designed retro-aldolases that employ four different catalytic motifs to catalyze the breaking of a carbon-carbon bond in a non-natural substrate. Designs that utilized an explicit water molecule to mediate proton shuffling were significantly more successful, with rate accelerations of up to four orders of magnitude and multiple turnovers, than those involving charged sidechain networks. The atomic accuracy of the design process was confirmed by the X-ray crystal structure of active designs embedded in two protein scaffolds, both of which were nearly superimposable on the design model.
Using new algorithms that employ hashing techniques to construct active sites for multi-step reactions, we designed retro-aldolases that employ four different catalytic motifs to catalyze the breaking of a carbon-carbon bond in a non-natural substrate. Designs that utilized an explicit water molecule to mediate proton shuffling were significantly more successful, with rate accelerations of up to four orders of magnitude and multiple turnovers, than those involving charged sidechain networks. The atomic accuracy of the design process was confirmed by the X-ray crystal structure of active designs embedded in two protein scaffolds, both of which were nearly superimposable on the design model.
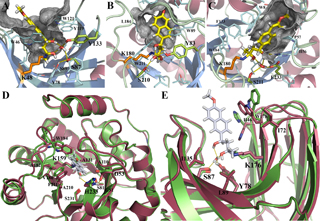
Structures of artificial retro-aldol enzymes
(A, B, C). Examples of design models for active designs highlighting groups important for catalysis. The nucleophilic imine-forming lysine is in orange, the transition state model is in yellow, hydrogen bonding groups are in light green, and the catalytic water is shown explicitly. The designed hydrophobic binding site for the aromatic portion of the transition state model is indicated by the gray mesh. (D, E). Overlay of design model (purple) on X-ray crystal structure (green).